Julia might just change the game for bioinformatics. Bioinformatics is starting to heat up as a field, and I think we have a lot of unique issues starting from the days of perl and the Human Genome Project.
Based off a recent recommendation from my friend Teco
Two Language Problem
Plenty of people have written about this with Julia. Lots of scientific communities may use a high-level "scripting" language to manage their business-logic and then drop into a low-level language such as C/Fortran/Rust to highly optimize the bottleneck.
In bioinformatics, we have a 5 language problem. From the perl script written before the first generation of Next Generation Sequencing, to packages like DESeq2 that aren't going to leave anyone's toolbox anytime soon, but no one is going to rewrite, and instead are going to spend time teaching R to every incoming generation of bioinformaticians just so they can use these influential packages. That's not including python, Rust, and every bioinformatician's favorite, bash. That's not including if you want to contribute to any GATK CLI tools written in Java.
Now, workflow managers such as Snakemake and Nextflow have fixed the majority of those. Just stick those scripts in a container that acts as a time-capsule of versions long forgotten, and you can use them for just what you need and put them back in closet.
But what about new scripts? When you're starting a new bioinformatics project in 2022, do you follow in the footsteps of the pioneers that came before you, and learn how to call C functions in an R package? Or would you rather just see this beauty.

But what about my C functions that are highly optimized?
What about our in-house python library that interacts with all of our sample tracking and pulling files temporarily from s3? "I don't have time to figure out how to untangle that mess" you say. The Julia community wants to keep all of those import scientific scripts written. Packages such as RCall.jl, PyCall.jl, and plenty of others housed under Julia Interop allow for legacy code to fit right into new code. As scientists, we are constantly standing on the shoulders of giants, and Julia is enabling that.
BioJulia
Julia has a wonderful open-source structure. Since it was created, only a decade ago there's not a lot of legacy things to maintain and everything can be fresh and inclusive. Take for example the heavy use of organizations to house these code repositories, which prevents packages from going unmaintained and orphaned when the creator moves on.
While it's a relatively small community, they've covered a wide range of the various file types that we deal with on a daily basis. I hope to dog food most of the packages to fill in some missing pieces, and improve documentation and create some content for the community. The part that I found difficult was finding the people based on the website. I joined the Gitter to ask a question, but luckily someone saw the message and told me that most of the real-time chat happens in the #biology channel in the Julia slack.
Designed for Scientific Computing
It seems like bioinformaticians always want to do things in the most efficient path, but solving for a different variable than most. In a recent episode of screaming in the cloud, Lynn Langit mentioned that in finance, they care about getting the results as quickly as possible. The cost of the computing isn't a factor. In bioinformatics, they're trying to solve for both time and cost, trying to find the local minimum between both. There are plenty of time results don't need to be instant, and waiting a few days to stretch a grant out is a necessary evil.
Julia is fast, and that's been said numerous times, so you're probably guessing I'm going to say you can save money by decreasing your compute time. While that's true, the piece that I think people aren't solving for in that equation is developer time. Julia was designed from the ground up as a general programming language for scientists (Why We Created Julia).
We want a language that's open source, with a liberal license. We want the speed
of C with the dynamism of Ruby. We want a language that's homoiconic, with true
macros like Lisp, but with obvious, familiar mathematical notation like Matlab.
We want something as usable for general programming as Python, as easy for
statistics as R, as natural for string processing as Perl, as powerful for
linear algebra as Matlab, as good at gluing programs together as the shell.
Something that is dirt simple to learn, yet keeps the most serious hackers
happy. We want it interactive and we want it compiled.
That sounds like a bioinformatician's dream to me! Think of all the time we can save on developer experience, not trying to hack out some extra speed, or fixing broken dependencies (or a complete lack of specified dependencies!). Allowing legacy code to be treated like the crown jewel in our metaphorical software crown, and wrapping it in some gold Julia code like it deserves.
Call to action
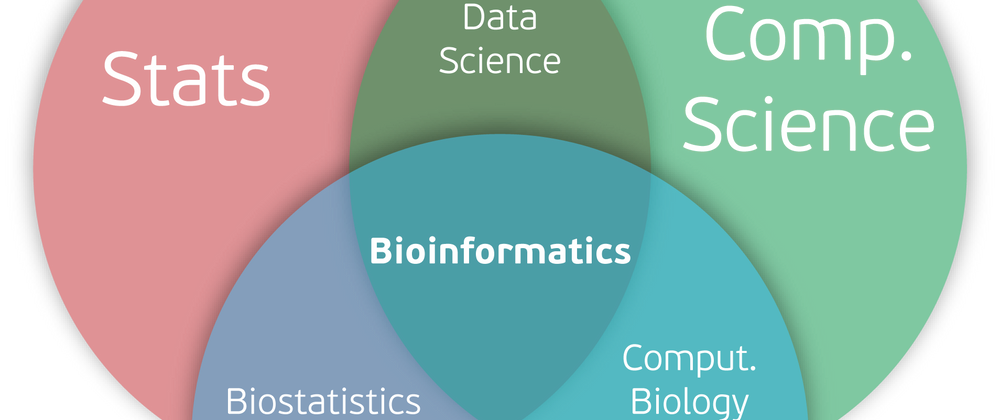
I plan on working to increase the visibility into Julia, specifically for bioinformatics. I always love this Venn diagram to explain to people what bioinformatics is. I think it'll be natural to cover JuliaData, JuliaStats, and BioJulia to cover all three and show people how the three intersect.
How to get started with Julia
- Attend JuliaCon 2022 (It's online and free to attend!)
- Listen to the Talk Julia Podcast
- Go through some courses on JuliaAcademy
- Check out the work by Logan Kilpatrick, the Julia Dev Community Advocate

Top comments (2)
This is a great post! I'm an active developer in the scope of Computational Chemistry (with a focus on protein design) and I must say: solving the two-language problem has brought the protein design software back to the 21st century! Eager to see what new development show up in this field!
This is a really great overview post! Please let me know if I can be of any help supporting the Bioinformatics Julia community!