
How do you rationally engineer the metabolism of a microorganism? First, you need to understand how it works of course! The blueprint describing how a cell functions is encoded by its genome. A cell's genome is the collection of all its genes, which are composed of DNA, and acts like a parts list. Each gene usually gets translated into an enzyme. These enzymes catalyze metabolic reactions, which are used to convert food (e.g. sugars like glucose) into energy, new cells, and other metabolic products, like biofuels. The reactions catalyzed by all these enzymes is what we call the cell's metabolism.
Over the past few decades synthetic biology has made amazing strides. We are now able to engineering the genome with exquisite precision. For example, it is possible to modify single DNA bases, cut out endogenous genes, insert entirely new genes, and artificially regulate the expression of enzymes with relative ease. An ongoing goal is to better understand the metabolism of an organism so that these gene editing technologies can be harnessed to design and engineer microbes like we would a car. This can be used to save the planet from fossil fuels!
However, to get there, we need to systematically understand how changes at the gene level impact metabolism. If we can do this, we will know which genes (parts) to modify to bend metabolism to our will. A popular mathematical framework to accomplish this is via constraint-based reconstruction and analysis (COBRA). Briefly, COBRA entails cataloging all the reactions encoded for by the genome of an organism, and constructing a genome-scale metabolic model. Typically, these metabolic models are composed of 1000s of reactions that describe what metabolites an organism is capable of producing. Importantly, through painstaking work, these reactions are linked to specific genes. Using an optimization based technique called flux balance analysis (FBA, described in detail later), it is possible to predict the rate at which metabolic enzymes work (carry flux). The gene reaction rules encoded by the model link specific reactions to genes, allowing us to predict what would happen if the genome of an organism were to be modified.
Recently, various packages, written purely in Julia, have been released to bring these kinds of analyses to our community. Will will use COBREXA and Escher to demonstrate how a toy model of Escherichia coli's central metabolism can be engineered to enhance its production of acetate and ethanol.
First, we need to download the genome-scale metabolic model and visualize it.
using COBREXA, Escher, CairoMakie, Tulip, Colors
# download a toy model of Escherichia coli's central metabolism
download("http://bigg.ucsd.edu/static/models/e_coli_core.json", "e_coli_core.json")
# download the metabolic map
download("http://bigg.ucsd.edu/escher_map_json/e_coli_core.Core%20metabolism", "e_coli_core_map.json")
# visualize the metabolism
escherplot(
"e_coli_core_map.json";
reaction_show_text = true,
reaction_edge_color = :grey,
metabolite_show_text = true,
metabolite_node_colors = Dict("glc__D_e" => :red),
metabolite_node_color = :lightskyblue,
)
hidexdecorations!(current_axis())
hideydecorations!(current_axis())
current_figure()
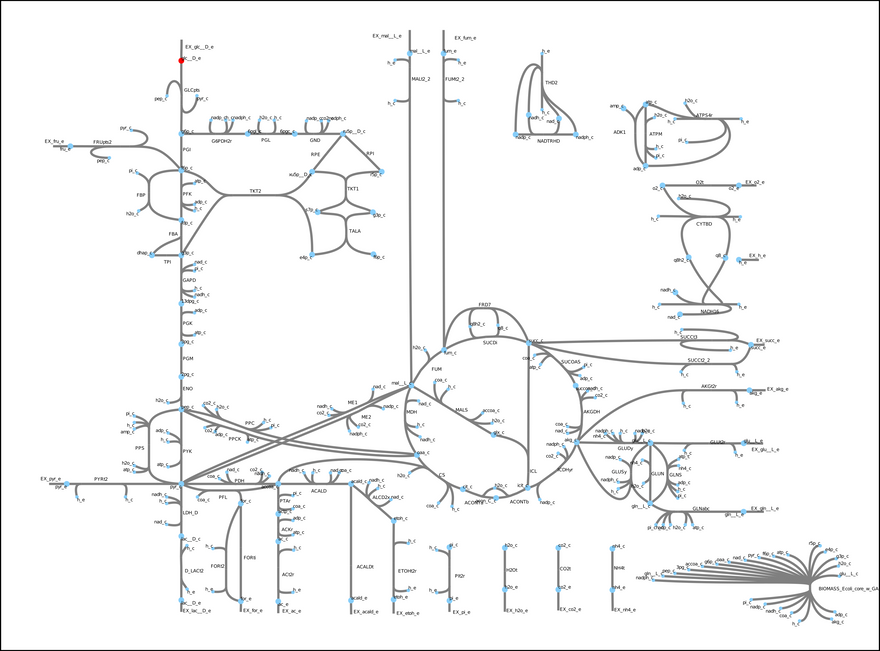
This model includes glycolysis, the TCA cycle, and the electron transport chain. These metabolic modules are important in the central carbon metabolism of most cells and organisms. In short, glucose (a sugar, highlighted in red) is converted into energy (ATP) and other metabolic precursors that are used to produce more biomass (cells). Each edge in the figure is a reaction, and each node a metabolite. Most of the reactions in this model actually exist in E. coli. One exception is the so-called biomass objective function (bottom right hand corner). Essentially, it is a pseudo-reaction that lumps all the unmodeled processes that convert the metabolites generated by the model into biomass. A fundamental assumption behind constraint-based models is that, through evolution, microbes have tuned their metabolism to satisfy some objective. In some cases, growth rate maximization is a good enough simplification.
This is where FBA comes into the picture. At its core, FBA formulates these genome-scale metabolic models as optimization problems:
Here, the metabolism is represented through the stoichiometric matrix, S, which enforces mass balance on the model. It acts to link reactions and metabolites within the model to each other. The biomass objective function is denoted μ, and is a linear function of the reaction fluxes, v, which themselves can be bounded based on measured data. While metabolic models tend to be really big and complex, FBA reduces to a linear program, meaning it can be solved efficiently!
By making use of Julia's excellent ecosystem, we were able to build COBREXA. It is a package for performing all manner of constraint-based modeling simulations and reconstructions. So, let's use it to engineer our toy model!
First, let's simulate the growth of E. coli under its default conditions (aerobic, glucose fed), and use COBREXA to predict its growth rate and active reactions with FBA.
# load the model
model = load_model("e_coli_core.json")
# perform flux balance analysis
fluxes = flux_balance_analysis_dict(model, Tulip.Optimizer)
which results in:
julia> flux_summary(fluxes)
Biomass
BIOMASS_Ecoli_core_w_GAM: 0.8739
Import
EX_o2_e: -21.7995
EX_glc__D_e: -10.0
EX_nh4_e: -4.7653
EX_pi_e: -3.2149
Export
EX_h_e: 17.5309
EX_co2_e: 22.8098
EX_h2o_e: 29.1758
Thus, our model predicts a growth rate of 0.87 1/h when the cell metabolizes 10 mmol glucose/gDW/h (the default input). This is pretty accurate compared to experimental results! We can easily visualize which reactions the cell uses under these conditions:
tol = 1e-3 # threshold if reaction is active
escherplot(
"e_coli_core_map.json";
reaction_edge_colors = Dict(id => :red for (id, flux) in fluxes if abs(flux) > tol),
)
hidexdecorations!(current_axis())
hideydecorations!(current_axis())
current_figure()
As you may see, only some of the reactions are used (the dashed ones are inactive). This is a realistic result, because microbes regulate their metabolism to suite their environmental needs. Specifically, the model uses respiration to produce energy, because it is a very efficient (you are doing it right now!).
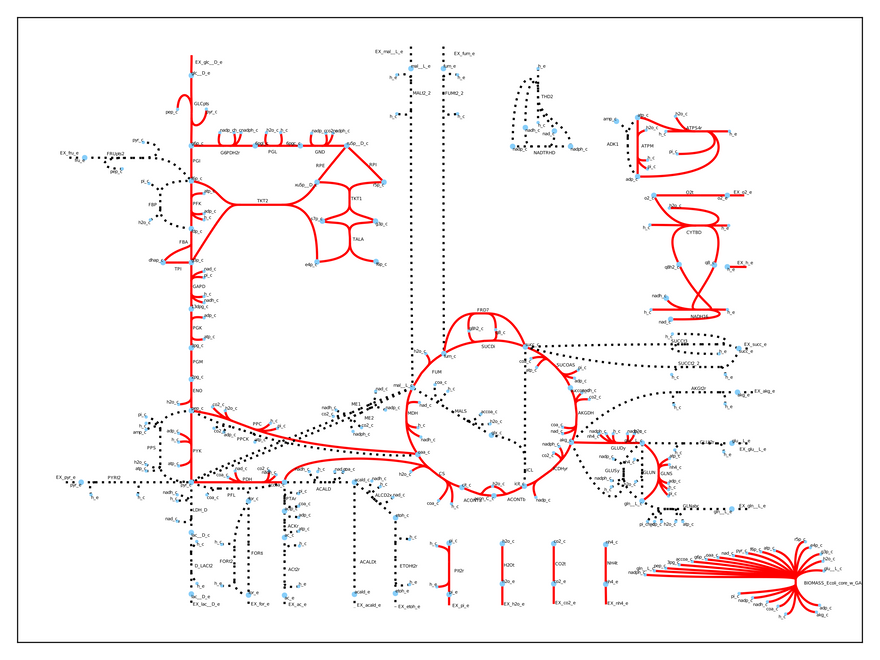
However, we would like our engineered microbe to produce ethanol to power our car. One way to achieve this is to knockout (delete) some genes to reroute cellular metabolism to achieve this goal. Let's delete the genes corresponding to cytochrome oxidase (one of the main reactions in aerobic respiration), and see what happens.
ko_fluxes = flux_balance_analysis_dict(
model,
Tulip.Optimizer;
modifications = [
knockout(["b0978", "b0734"]), # these genes code for cytochrome oxidase
],
)
which results in:
julia> flux_summary(ko_fluxes)
Biomass
BIOMASS_Ecoli_core_w_GAM: 0.2117
Import
EX_glc__D_e: -10.0
EX_h2o_e: -7.1158
EX_nh4_e: -1.1542
EX_pi_e: -0.7786
EX_co2_e: -0.3782
Export
EX_etoh_e: 8.2795
EX_ac_e: 8.5036
EX_for_e: 17.8047
EX_h_e: 30.5542
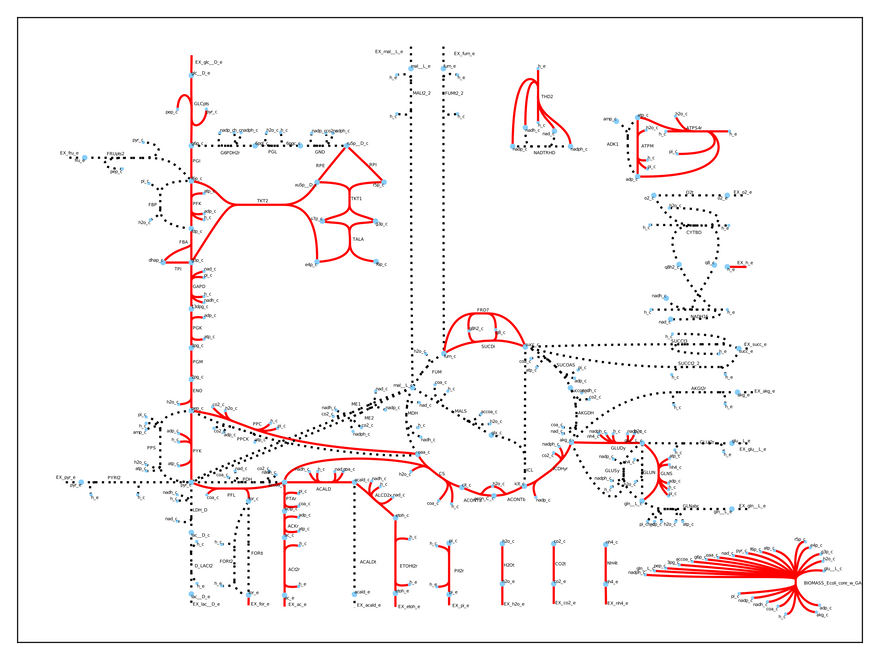
Whoa! We have totally changed the cell's physiology! Now, it is growing much slower (0.21 1/h), but it is producing both ethanol and acetate. We can visually inspect what is going again:
escherplot(
"e_coli_core_map.json";
reaction_edge_colors = Dict(id => :red for (id, flux) in ko_fluxes if abs(flux) > tol),
metabolite_node_color = :lightskyblue,
reaction_show_text = true,
metabolite_show_text = true,
)
hidexdecorations!(current_axis())
hideydecorations!(current_axis())
current_figure()
The entire metabolism has changed. Without these two crucial genes the cell uses fermentation to grow and we can use its metabolic byproducts (acetate and ethanol) to power our car.
COBREXA can do much more than these simple simulations, and was specifically designed to be scalable to massive systems of interacting microbes. You can read the docs here to see what else it can do. As a challenge, see if you can use the functions in COBREXA to identify which genes to delete to maximize the production of ethanol :)

Top comments (1)
I used cobra back when it was a matlab library for my master thesis. It is amazing to see it ported to Julia. I again use LP and MIP in my work nowadays just in different domain: youtu.be/IYae_sfzKMs